.pdf+-+SumatraPDF_2012-12-20_04-04-08.png) |
| 1,1,2-trichloroethane |
The ratio of the integrals make sense, since there are two protons in one environment and one in the other.
The relative shifts should make sense too, since the CH is attached to two chloride atoms, deshielding it more than the CH
2 which is attached to just one.
The signal splitting can be explained by the
n+1 rule. Each type of proton environment "senses" the number of equivalent protons,
n, which are three bonds away. That environment is then split into n+1 peaks. This process is also called splin-splin splitting, or coupling, or spin-spin coupling.
So in the above specta, the CH is coupled to two CH
2 protons, so it split into three peaks, or a triplet. The CH
2 environment is coupled to one CH proton, so it split into two peaks, or a doublet.
Coupling does not happen between protons in the same environment. This is why the NMR of CH
4 would give a single peak.
Two more examples are below, ethyl iodide:
And 2-nitropropane:
.pdf+-+SumatraPDF_2012-12-20_04-23-50.png) |
| 2-nitropropane |
You might wonder why coupling only works within three bonds. That isn't entirely true, it is just that the coupling effect falls rapidly with distance. So except in special cases, such as double and aromatic bonds, coupling further than three bonds will not be visible on spectra. It would be visible if we had higher resolution machines.
Consider 1-propanol:
The n+1 rule tells us that both the CH
3 and the CH
2O group will split into 3 peaks. Coupling does not happen to the OH proton since it is rapidly exchanged with the solvent, so it has an average spin of 0, and experiences an average coupling of 0.
For the central CH
2 group, there are two different sets of neighboring equivalent protons, so we have to use the n+1 rule twice, one for splitting by the CH
2O and once for splitting by the CH
3. The result is a quadruplet of triplets, or a triplet of quadruplets, giving us 12 predicted peaks in total:
In practice, high number of peaks are very hard to distinguish. A peak like the one at 1.5 ppm above would just be called a "multiplet", at least at undergrad level.
Remember that coupling only works between non equivalent protons. So HA and HB below would give a singlet.
.pdf+-+SumatraPDF_2012-12-31_13-19-40.png)
.pdf+-+SumatraPDF_2012-12-31_06-42-32.png)
.pdf+-+SumatraPDF_2012-12-31_06-28-43.png)
.pdf+-+SumatraPDF_2012-12-31_06-06-40.png)
.pdf+-+SumatraPDF_2012-12-31_05-56-18.png)
.pdf+-+SumatraPDF_2012-12-26_04-37-49.png)
.pdf+-+SumatraPDF_2012-12-26_03-18-08.png)
.pdf+-+SumatraPDF_2012-12-26_01-14-44.png)
.pdf+-+SumatraPDF_2012-12-22_09-30-33.png)
.pdf+-+SumatraPDF_2012-12-22_02-05-06.png)
.pdf+-+SumatraPDF_2012-12-22_01-32-38.png)
.pdf+-+SumatraPDF_2012-12-22_01-19-03.png)
.pdf+-+SumatraPDF_2012-12-21_20-55-11.png)
.pdf+-+SumatraPDF_2012-12-21_20-59-46.png)
.pdf+-+SumatraPDF_2012-12-21_15-05-36.png)
.pdf+-+SumatraPDF_2012-12-21_08-24-05.png)
.pdf+-+SumatraPDF_2012-12-21_08-58-25.png)
.pdf+-+SumatraPDF_2012-12-21_09-00-55.png)
.pdf+-+SumatraPDF_2012-12-21_09-01-47.png)
.pdf+-+SumatraPDF_2012-12-21_07-17-00.png)
.pdf+-+SumatraPDF_2012-12-21_07-18-30.png)

.pdf+-+SumatraPDF_2012-12-21_04-25-12.png)
.pdf+-+SumatraPDF_2012-12-21_04-25-23.png)
.pdf+-+SumatraPDF_2012-12-21_05-03-49.png)
.pdf+-+SumatraPDF_2012-12-21_02-05-03.png)
.pdf+-+SumatraPDF_2012-12-21_02-11-46.png)
.pdf+-+SumatraPDF_2012-12-21_02-56-26.png)
.pdf+-+SumatraPDF_2012-12-20_04-04-08.png)
.pdf+-+SumatraPDF_2012-12-20_04-21-57.png)
.pdf+-+SumatraPDF_2012-12-20_04-22-09.png)
.pdf+-+SumatraPDF_2012-12-20_04-23-33.png)
.pdf+-+SumatraPDF_2012-12-20_04-23-50.png)

.pdf+-+SumatraPDF_2012-12-21_02-56-34.png)
.pdf+-+SumatraPDF_2012-12-20_00-45-54.png)
.pdf+-+SumatraPDF_2012-12-20_00-47-58.png)
.pdf+-+SumatraPDF_2012-12-20_00-50-32.png)
.pdf+-+SumatraPDF_2012-12-20_00-50-05.png)
.pdf+-+SumatraPDF_2012-12-20_00-50-21.png)
.pdf+-+SumatraPDF_2012-12-20_01-04-21.png)
.pdf+-+SumatraPDF_2012-12-20_01-06-29.png)
.pdf+-+SumatraPDF_2012-12-19_21-20-25.png)
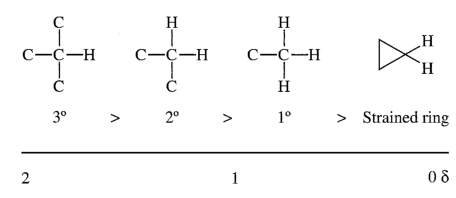.pdf+-+SumatraPDF_2012-12-19_21-31-36.png)
.pdf+-+SumatraPDF_2012-12-19_08-01-34.png)
.pdf+-+SumatraPDF_2012-12-19_07-57-33.png)
.pdf+-+SumatraPDF_2012-12-19_07-53-58.png)
.pdf+-+SumatraPDF_2012-12-19_07-22-27.png)
.pdf+-+SumatraPDF_2012-12-19_06-55-30.png)
.pdf+-+SumatraPDF_2012-12-19_06-55-38.png)